Posteriormente, se identificaron varias familias en las que los resultados no eran benignos, tenían convulsiones persistentes o que no respondían a las drogas, deterioro del desarrollo o ambos. Esto llevó a un grupo de investigadores (entre los que encontramos a la muy relevante Dra Sarah Weckhuysen) a examinar a los pacientes con síndromes de epilepsia neonatal graves para detectar mutaciones en KCNQ2.
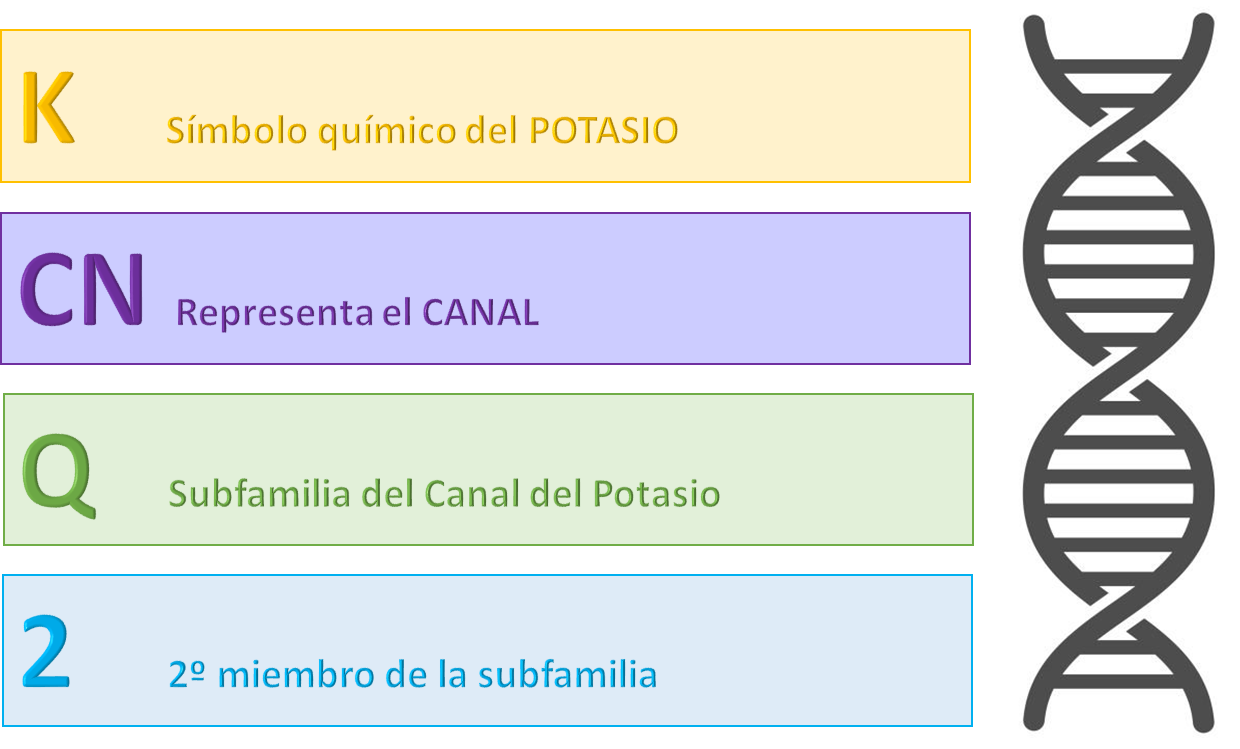
¿Qué es el gen KCNQ2?
El Gen KCNQ2 codifica una proteína que forma parte de los canales de potasio en las neuronas, y las mutaciones de este gen pueden afectar a la función de estos canales, causando excitabilidad neuronal excesiva y convulsiones.
La historia de la identificación de la encefalopatía epiléptica por mutación en KCNQ2 comienza con la identificación y caracterización de la epilepsia neonatal familiar benigna (BFNE).
Esta condición fue descrita por primera vez como un síndrome en 1964 por los médicos Rett y Teubel.
Informaron de una familia con ocho personas afectadas durante 3 generaciones. El niño más pequeño tuvo convulsiones a los 3 días de edad descritas como eventos tónico-clónicos que ocurren varias veces al día. Los EEG fueron normales entre las convulsiones y los niños se desarrollaron adecuadamente después de que las convulsiones remitieron. Por lo general, esto sucedía más tarde en la infancia. Durante los siguientes veinte años, se describieron otras familias con historias similares. En algunos casos, las convulsiones persistieron hasta la vejez, pero los resultados fueron favorables.
En 1998, los investigadores identificaron un gen en la región que parecía similar en estructura a un canal de potasio dentro del corazón. Este nuevo gen se ha denominado convencionalmente KCNQ2
Se identificaron ocho casos de ese grupo de 80 pacientes, y esos niños compartían muchas características. Desde 2011, se han diagnosticado a muchas más personas y el síndrome se ha definido con más detalle.
Encefalopatía epiléptica por KCNQ2
La Epilepsia Neonatal Familiar Benigna (BFNS) no entraña más complicación que en el nacimiento del bebé. Aparecen crisis epilépticas en las primeras horas/días de vida y desaparecen entre el primer y cuarto mes.
Lo que nos ocupa principalmente en la Fundación KCNQ2 España, es la Encefalopatía Epiléptica KCNQ2. Esta es la forma más grave de la alteración que provoca crisis epilépticas difíciles de controlar y van de la mano de una discapacidad intelectual y motora.
La mayoría de los casos de la Epilepsia Neonatal Familiar Benigna (BFNS) es hereditaria, mientras que la mayoría de los casos de la Encefalopatía Epiléptica por mutación en KCNQ2, se dice que es de NOVO. Esto se refiere a una alteración genética que aparece por primera vez en un individuo y no se ha heredado de sus padres ni está presente en generaciones anteriores de la familia. Este tipo de mutación surge en una célula germinal (óvulo o espermatozoide) de uno de los progenitores o directamente en el cigoto durante las primeras etapas del desarrollo embrionario.
La encefalopatía epiléptica se caracteriza por la aparición temprana de crisis epilépticas de muy diversos tipos, acompañadas de deterioro cognitivo y conductual, causado en parte, por la propia actividad epiléptica. Suelen ser resistentes al tratamiento farmacológico y pueden afectar significativamente el desarrollo neurológico. Estas son las características principales:
No existe todavía un tratamiento específico que corrija la encefalopatía epiléptica por KCNQ2, pero si hay diferentes opciones de medicamentos, para tratar las convulsiones y otros efectos consecuencia de su situación, como son la espasticidad, trastornos del movimiento, etc.
Las convulsiones suelen aparecer en las primeras horas/días del recién nacido
En muchos casos, pueden ser difíciles de controlar con medicamentos
Afecta negativamente al desarrollo cognitivo, motor y del comportamiento
Los electroencefalogramas son anormales, mostrando patrones de actividad cerebral anormales
Diagnóstico de KCNQ2
Cuando en un bebé aparecen las crisis epilépticas, los médicos y aseguradoras buscan de lo más general a lo más específico. Es por esto que lo primero que hacen no son las pruebas genéticas. Primero hacen valoraciones por infecciones, alteraciones electrolíticas, metabólicas o problemas estructurales en el cerebro.
Se está viendo que es más frecuente y menos “raro” que pueda ser una alteración genética y por esto estas pruebas se hacen cada vez antes, siendo de gran importancia disponer de un diagnostico cuanto antes. El diagnóstico precoz y aplicación de fármacos ad hoc para KCNQ2, va a marcar el desarrollo del paciente KCNQ2.
La única forma actualmente de determinar el diagnostico de KCNQ2, es mediante pruebas genéticas moleculares. Se realiza mediante análisis del gen del canal de potasio o mediante una prueba genética que busque mutaciones en varios genes asociados con la epilepsia infantil.
Lo primero que sucede ante las convulsiones de un bebé, es que le someten a un Electro Encefalograma (EEG) para evaluar las crisis epilépticas y los patrones de actividad cerebral asociados a las mismas. Se colocan unos electrodos en el cuero cabelludo y registran las ondas eléctricas durante los periodos de actividad, sueño y convulsiones.
El encefalograma en el Encefalopatía epiléptica por KCNQ2 mientras no se suceden las crisis, normalmente NO es normal, mientras que en la Epilepsia Neonatal Familiar Benigna (BFNS), sí que tiende a ser normal.
La resonancia magnética (IRM) genera imágenes detalladas del cerebro utilizando un campo magnético. Las imágenes pueden proporcionar información sobre cualquier malformación de las estructuras cerebrales u otros tipos de lesiones que se ven comúnmente en la epilepsia. Normalmente la resonancia magnética de un paciente con Encefalopatía epiléptica causada por KCNQ2, nos dicen que es normal. A veces sus malformaciones son demasiado pequeñas para ser detectadas con una resonancia magnética.
¿Existen las terapias para tratar el KCNQ2?
Hasta que un paciente recibe su diagnóstico de KCNQ2, tal y como nos han dicho en muchas ocasiones, los médicos utilizan diferentes recursos de fármacos de los más frecuentes a los menos, tratando de controlar las convulsiones.
Una vez se recibe el diagnóstico, los fármacos a utilizar son mucho más dirigidos, sabiendo los que funcionan mejor para estos casos y aplicándolos. El uso de estos fármacos, puede ayudar a controlar o solamente reducir las crisis epilépticas asociadas con KCNQ2. Actualmente se intenta dirigir a los pacientes a un tratamiento farmacológico más individualizado, en función de la mutación que porta.
Estos fármacos no siempre funcionan de la misma forma en cada paciente. Si el paciente no responde a tratamiento farmacológico, a veces se consideran otros tratamientos como dietas cetogénicas, dispositivos o cirugías especializadas.
Por tanto, el abordaje de la enfermedad es multidisciplinar, involucrando, no sólo al equipo neurológico, sino también genétistas, rehabilitadores y fisioterapeutas, terapeutas ocupacionales, logopedas, psicólogos…
Como adicional, muchos KCNQ2 son diagnosticados a su vez como autistas como resultado de los daños de la alteración genética. Esto se debe a su comportamiento con movimientos repetitivos, falta de integración sensorial, contacto visual deficiente, etc. Es por ello que, en estos pacientes, las terapias para tratar el autismo pueden ser de gran utilidad.
")
")
")



















")
")














")

")
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Terapia génica
Actualmente, no existe una terapia génica estándar para las enfermedades relacionadas con el gen KCNQ2, aunque se está investigando activamente y se han reportado avances prometedores en ensayos clínicos y proyectos de investigación.
El tratamiento principal para los niños con KCNQ2 es sintomático, centrado en el control de las convulsiones con medicamentos anticonvulsivos, como los bloqueadores de los canales de sodio (carbamazepina, fenitoína), los cuales se han mostrado eficaces en algunos pacientes, especialmente en la encefalopatía por KCNQ2.
Aunque todavía en fases de investigación, la terapia génica representa una esperanza a largo plazo para corregir las mutaciones subyacentes. Los enfoques de terapia génica incluyen la adición génica, el silenciamiento génico y la corrección génica.
Hospitales como Sant Joan de Déu y el Hospital Infantil de Filadelfia son centros activos en la investigación de estas enfermedades y la búsqueda de nuevas estrategias terapéuticas, incluida la terapia génica.
En la terapia génica utilizan la transferencia de material genético para prevenir o curar enfermedades genéticas. Es la mejor de las opciones, pero también la más difícil. Va directamente a la raíz del problema mediante la transferencia de la versión correcta de un gen defectuoso, que es el que está causando la enfermedad.
Se puede conseguir restablecer la función del gen mutado, y la estrategia más común es la introducción de una copia normal de éste en las células.
La terapia génica puede ser aplicada mediante diferentes técnicas:
La terapia génica puede ser aplicada mediante diferentes técnicas:
- Terapia Génica Somática
- Terapia Germinal: sobre células germinales, que serían los espermatozoides y los óvulos. Esta se ha objetado que viola la dignidad humana al cambiar el contenido genético de las siguientes generaciones.
Dentro de las Terapias Génicas Somáticas, se pueden aplicar diferentes técnicas:
- Terapia Ex Vivo: Extraer células a reparar, repararlas en laboratorio y reimplantarlas en el organismo del paciente.
- Terapia In Situ: Introducir el gen repararador en el organismo del paciente
- Terapia In Vivo: Administrar el gen corrector al paciente
Estas son las terapias que nos hacen vibrar y luchar por conseguir llegar a aplicarlas en nuestros pacientes KCNQ2. Hacia aquí van todos nuestros esfuerzos y por esto estamos aquí, por lograr el máximo para nuestros pacientes.
Pedimos investigación para terapia génica aplicada a KCNQ2 ya!!